Sustainable Polymers for Packaging Applications
|
Since 2001, the Miller Research Group has worked in a variety of areas: synthetic organic chemistry, physical organic chemistry, organometallic chemistry, inorganic chemistry, industrial chemistry, polymerization chemistry, polymer science, environmental chemistry, and theoretical chemistry. Over time, our research has increasingly focused on polymerization chemistry and, more specifically, the realm of sustainable, environmentally-friendly polymers. The over-arching goal of our research has been to design, synthesize, and characterize new biogenic polymers that have the potential to replace incumbent packaging plastics, which are fossil fuel-based polymers with slow degradation behaviors. Thus, from plant-based building blocks, we aim to synthesize new polymers that degrade readily and benignly in the environment.
|
|
Water-degradable polymers
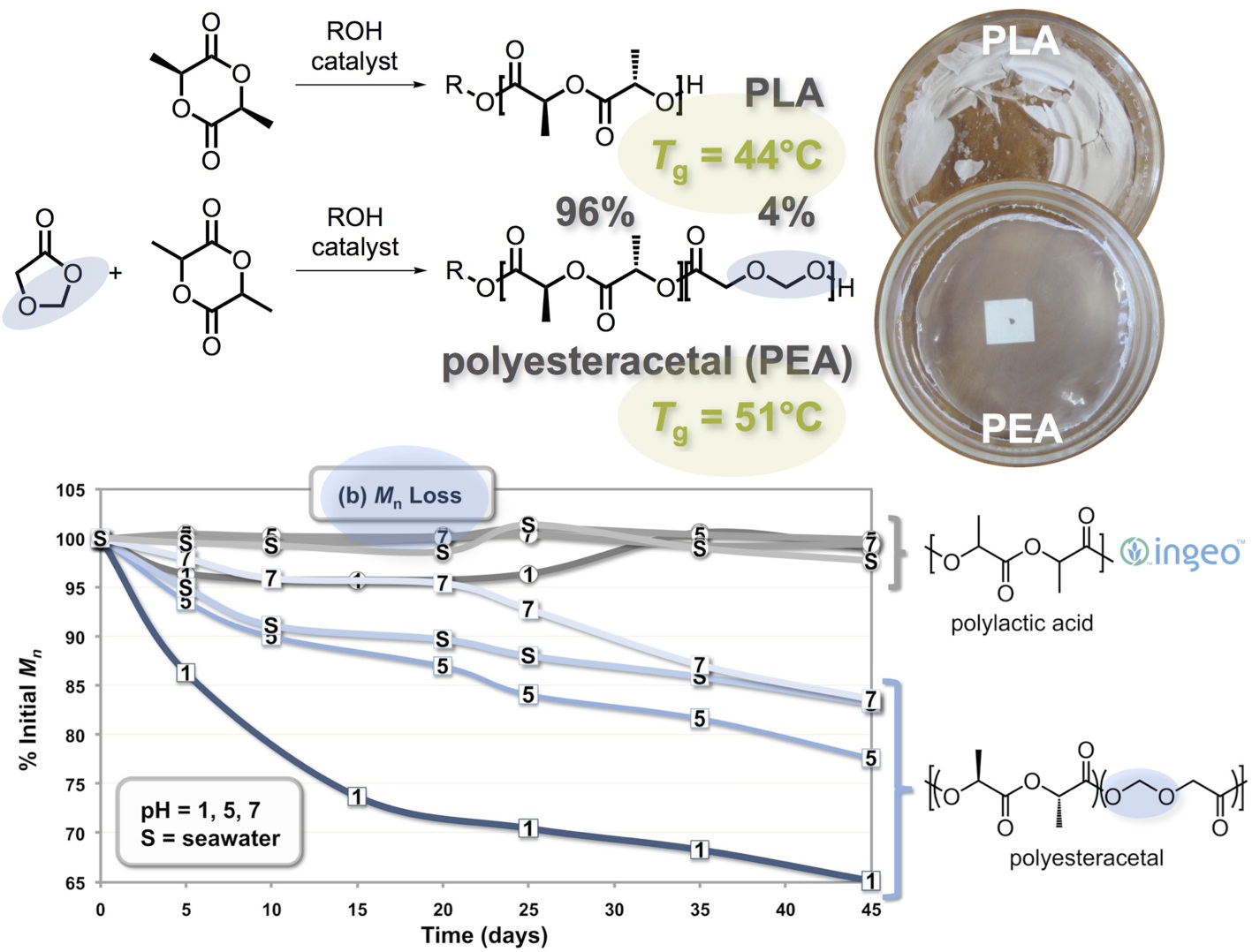
Our research efforts have been strongly inspired by Nature. For example, the most abundant polymer on earth is cellulose, which harbors half of all organic carbon. Each monomer is connected by an acetal functional group, but this functionality is nearly absent in the commercial polymer repertoire. We have incorporated the acetal functional group into the main-chain of polylactic acid (PLA), generating a family of polyesteracetal (PEA) copolymers. [1,2] The ring-opening copolymerization of lactide with 1,3-dioxolan-4-one (DOX, a cyclic ester acetal, approximately 4% incorporation) yields a polyesteracetal, a class of polymers that is very minimally explored. This material is essentially a copolymer of polylactic acid, glycolic acid, and formaldehyde, although it appears that higher polymerization temperatures sometimes effect formaldehyde extrusion, rendering isolated glycolate segments. The DOX comonomer can be industrially produced in >99% yield from trioxane and carbon monoxide, or in principle from wood alcohol (as methanol was once called when we obtained it exclusively via wood distillation). Compared to PLA, the copolymer is tougher, stronger, quieter, more flexible, less brittle, more optically transparent, and it has a higher glass transition temperature. Another important advantage is that it degrades abiotically—that is, it does not require composting conditions. Unlike PLA, it will degrade at the bottom of a landfill through acid-catalyzed hydrolysis of the acetal group. It will even degrade in seawater and our extrapolations suggest that it will degrade fully in 5 to 10 years in the ocean; PLA itself shows no such degradation in seawater during our 45-day experiments. Initial target applications include disposable blister packaging and film applications, such as grocery bags.
|
|
Functional group metathesis polymerization
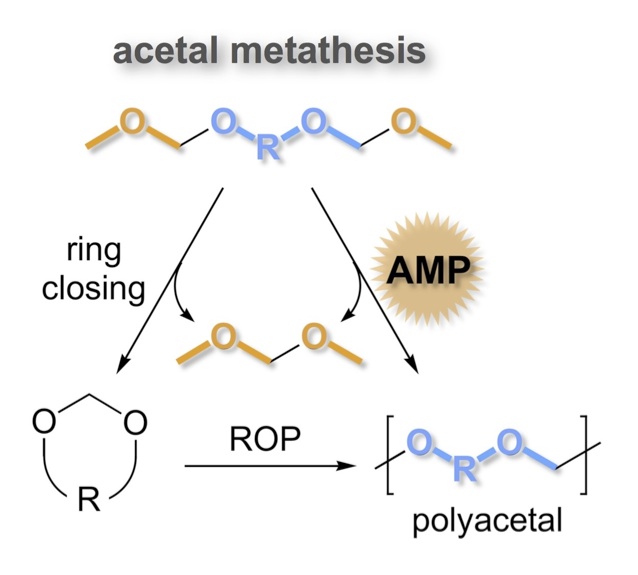
Olefin metathesis polymerization is an established route for converting olefins into polymers with an all-carbon main-chain. We have applied the principles of Acyclic Diene Metathesis (ADMET) polymerization to the development of an analogous methodology that converts acyclic bis-acetals into high molecular weight polyacetals. This Acetal Metathesis Polymerization (AMP) approach is a general method for converting diols—especially those from biorenewable triglycerides—into polymers particularly degradable via acid-catalyzed hydrolysis. [3,4] Thus, the polymers have sustainable origins and are designed to degrade even in the absence of microbial action (abiotic degradation).
 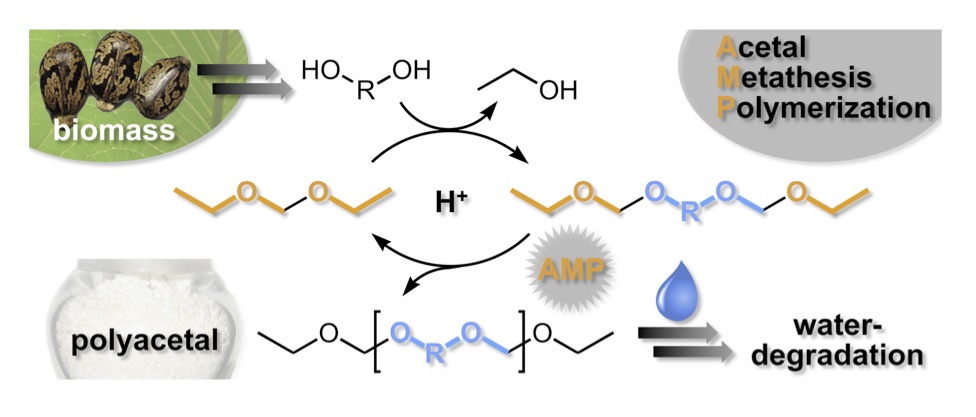 
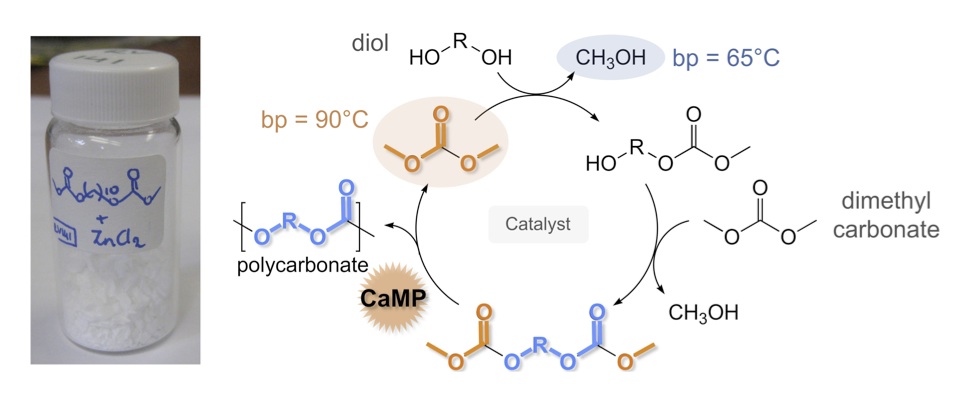
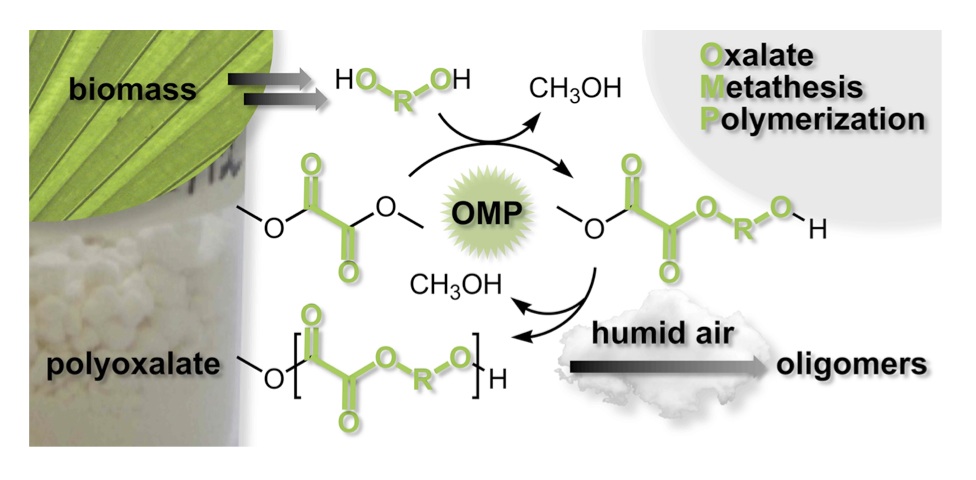
The AMP process is just one of several generalized functional group metathesis polymerization methods that our group has successfully applied to other functional groups, yielding a variety of novel polymeric structures derived from sustainable monomers—but designed to replace petroleum-based plastics. Other amenable functional groups include: carbonates, oxalates, and silicon acetals for the preparation of polycarbonates, polyoxalates, and polysilicon acetals, respectively. Carbonate Metathesis Polymerization (CaMP) was developed and applied to form high molecular weight polycarbonates (Mn up to 49,400) from renewable diols and dimethyl carbonate. [6] The CaMP methodology can be applied in a stepwise fashion that first isolates bis-carbonates or it can be applied directly to diols. A survey of twenty catalysts was conducted and K2CO3 was identified as competent for the direct method since it catalyzes both carbonate formation from alcohols and carbonate interchange. Diol selection and copolymerization strategies—including the incorporation of aromatic diols—allowed for precise tuning of polymeric thermal properties. Oxalate Metathesis Polymerization (OMP) was developed for the synthesis of polyoxalates. [7] This methodology employed para-toluene sulfonic acid for the acid-catalyzed ester interchange of dimethyl oxalate and linear diols HO–(CH2)n–OH (n = 3 – 12). Poly (decylene oxalate), a known polymer based on (potentially biorenewable) decanediol was synthesized in 83% yield with this methodology and exhibited no discernable Tg, a Tm of 79 °C, and a Mw of 67,600 g/mol. Polyarylene oxalates were derived from dimethyl oxalate and resorcinol bis(hydroxyethyl)ether or hydroquinone bis(hydroxyethyl)ether, aromatic diols that can be obtained from biorenewable resources. The poly(resorcinol bis(hydroxyethyl)ether) oxalate showed a Tg of 34 °C and a Tm of 156 °C. The poly(hydroquinone bis(hydroxyethyl)ether) oxalate showed a Tg of 45 °C and a Tm of 190 °C. Aliphatic/aromatic polyoxalate copolymers in varying compositions were prepared and studied. Incorporation of the aromatic diols into the polymer chain generally afforded increased Tg and Tm. Solid-state degradation studies indicated facile, abiotic water-degradation of the polyoxalates as molecular weights decreased 81–92% over the course of 13 months in humid air.
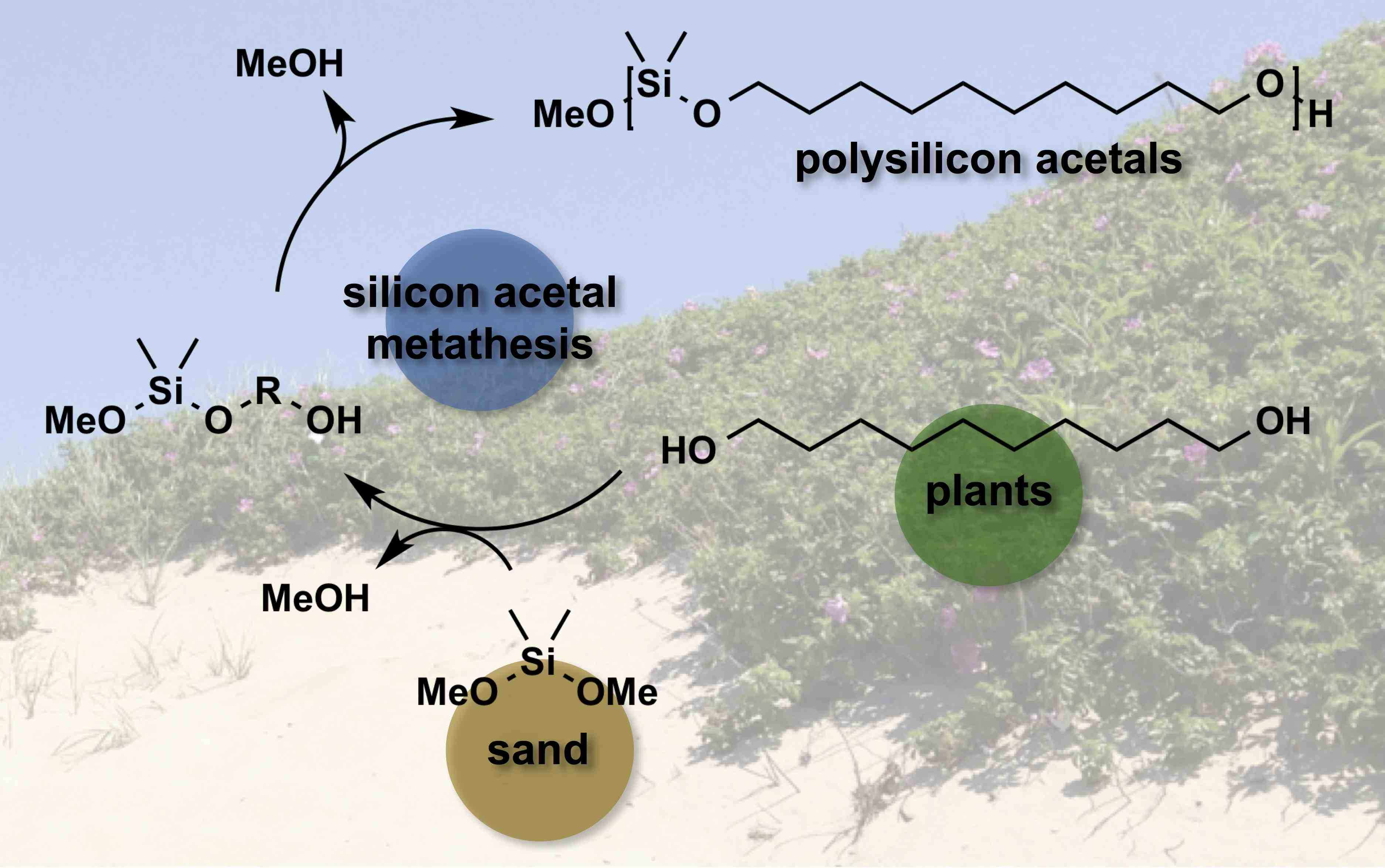
Both sustainable and non-sustainable diols HO–R–OH were employed to synthesize a variety of polysilicon acetals –[SiMe2O–R–O]– with tunable thermal properties via a convenient Silicon acetal Metathesis Polymerization (SAMP) route. [8] SAMP was accomplished as an acid-catalyzed silicon acetal interchange reaction between a diol and Me2Si(OR)2 (R = Me or Et) with 1:1 stoichiometry, driven by vacuum removal of volatile methanol or ethanol. This constitutes an efficient and general route to polysilicon acetals derived from diols and silicon, the second most abundant element in the Earth’s crust. Silicon acetal metathesis seems to be an underdeveloped oxygen-silicon bond forming reaction, certainly for the preparation of synthetic polymers. Ongoing studies seek a better understanding of the oxygen-silicon bond—-the most prevalent bond in our terrestrial environment—-in the context of synthesizing and degrading useful thermoplastics.
|
|
Polymers from C1 feedstocks
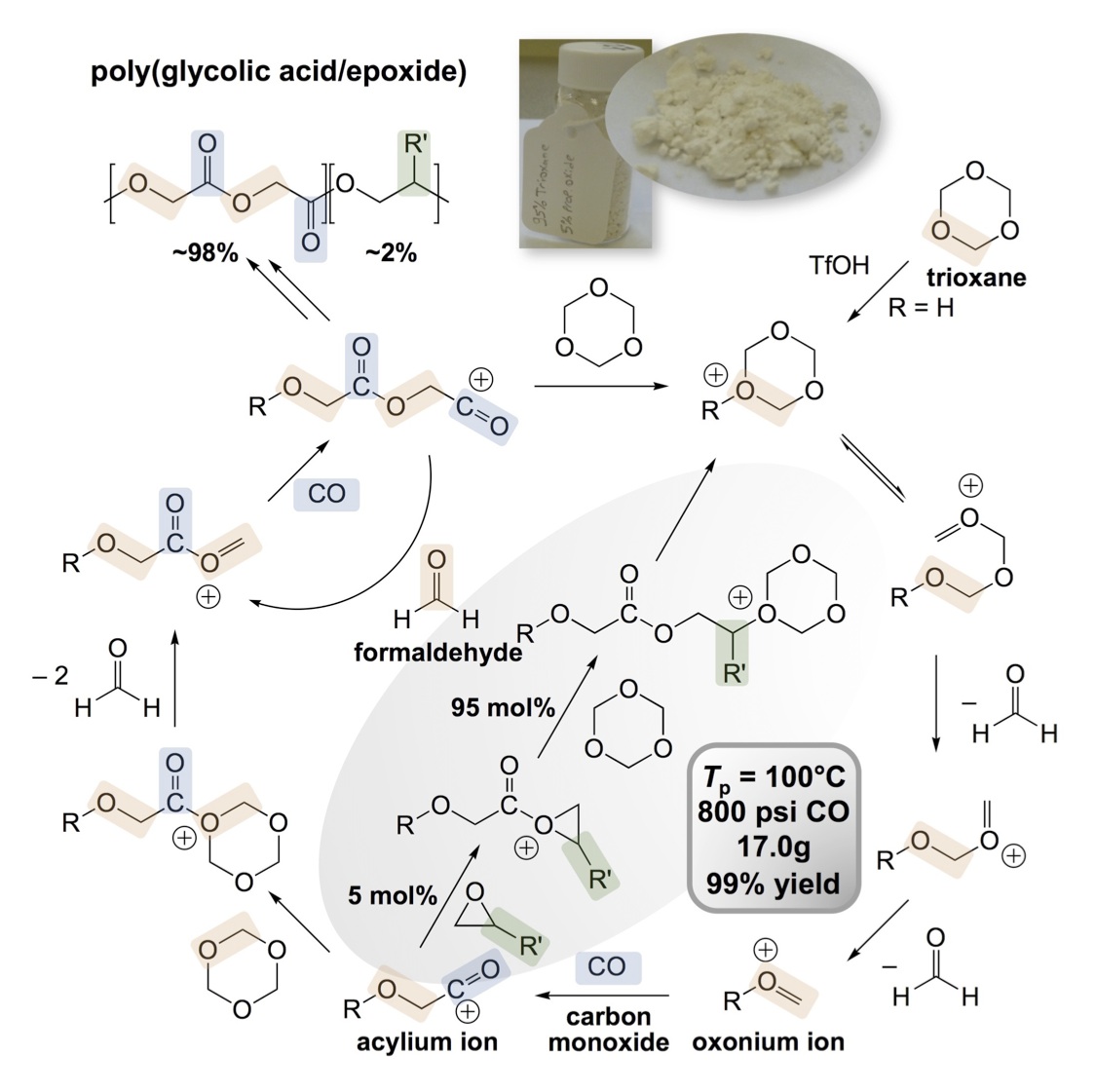
This research project targets the preparation of polymers from potentially biogenic C1 feedstocks despite the history that C1 feedstocks rarely function as monomers. We refined an approach to synthesizing polyglycolic acid (PGA) via the cationic alternating copolymerization of formaldehyde (from trioxane) and carbon monoxide (CO), sustainable C1 feedstocks obtainable from biomethanol, biogas, wood (wood alcohol), or non-food agricultural wastes. [9] This method constitutes an inexpensive and efficient pathway for the synthesis of PGA, circumventing the usual route requiring glycolide. PGA was successfully synthesized with yields up to 92% from trioxane, 800 psi of CO, and 1 mol% triflic acid (TfOH) initiator at 170 °C over three days. 1H NMR, 13C NMR, and FT-IR spectra of the polymer from CO and trioxane are identical to those of commercial PGA prepared via the ring-opening polymerization of glycolide—confirming the alternating microstructure. Although high copolymerization conversions were obtained, molecular weight analysis often suggested the formation of oligomeric glycolic acid. High molecular weight PGA can be obtained via post-polymerization polycondensation of the oligomers catalyzed by Zn(OAc)2·2H2O. Alternatively, increased molecular weight PGA can be achieved by inclusion of glycerol as a branching agent during the C1 copolymerization. Moreover, the polymerization method is readily amenable to the incorporation of comonomers, allowing for the facile modulation of the thermomechanical properties. Typical comonomers are inexpensive epoxides, which serve to disrupt the structural regularity of the polymeric main-chain, and thus will decrease crystallinity, modulus, and brittleness. We have prepared several examples of these PGA copolymers and continue to explore biogenic epoxide comonomers, such as those readily sourced from vegetable oils (triglycerides). We desire a full understanding of polymerizable comonomers and their effect on the polymeric properties.
|
|
Commodity thermoplastics from bioaromatics
Our polydihydroferulic acid (PHFA, termed GatoresinTM), [10,11] is derived from sustainable biomass feedstocks such as lignin-based vanillin and acetic acid and, importantly, mimics the structure and thermal properties of polyethylene terephthalate (PET) but does not rely on fossil fuels for its base feedstocks. [12] The glass transition temperature is 73°C and the melting temperature is 234°C, compared to values of 67 °C and 265 °C for PET. Moreover, the polymer is designed to degrade benignly into the environment either chemically (at the bottom of a landfill) or enzymatically (biodegradation under composting conditions) and the dihydroferulic acid building block is a metabolite recognizable by existing microbes. Initial target applications include beverage bottles, retail food containers, and eco-friendly soft textiles. We have accomplished the synthesis of polydihydroferulic acid from an alternate feedstock, ferulic acid, a naturally occurring hydroxycinnamic acid possessing antioxidant properties and abundantly available from the lignocellulose of megacrop agricultural wastes corn stover and sugarcane bagasse. It is the starting material for the synthesis of acetylferulic acid and its hydrogenated analogue, acetyldihydroferulic acid. These monomers, when copolymerized at various feed ratios, produce copolymers with tunable thermal and physical properties. [13] Some copolymers exhibit thermal properties comparable to commercially available fossil fuel-based packaging plastics—particularly polyethylene terephthalate (PET) and polystyrene (PS). For example, the copoly(ferulic acid/dihydroferulic acid) prepared with a 50:50 ratio of monomers has a Tg of 98 °C, an excellent match for that of polystyrene (PS, 100 °C). The glass transition temperature can be tuned from 78 °C to 153 °C and increases with increasing ferulic acid content and decreasing dihydroferulic acid content.
 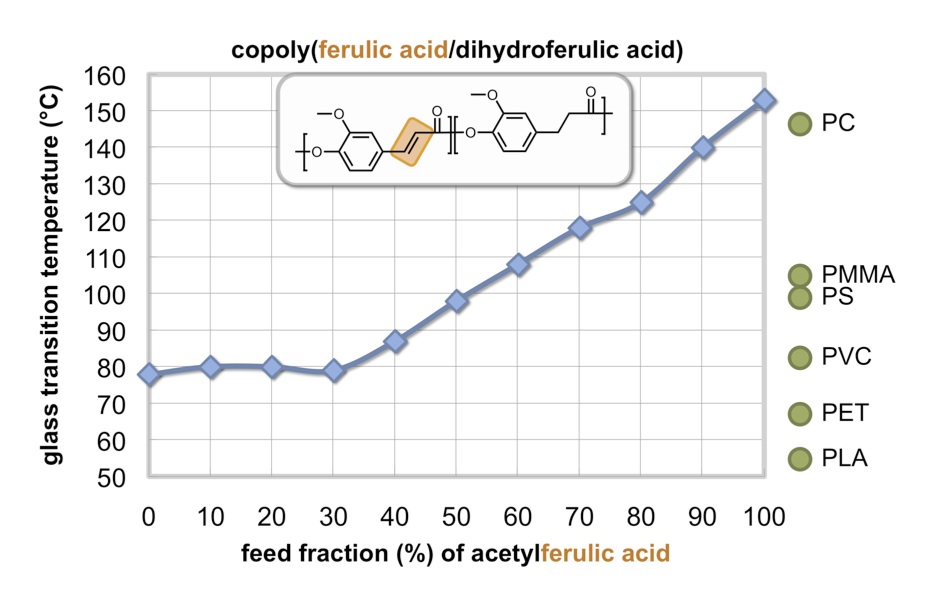 
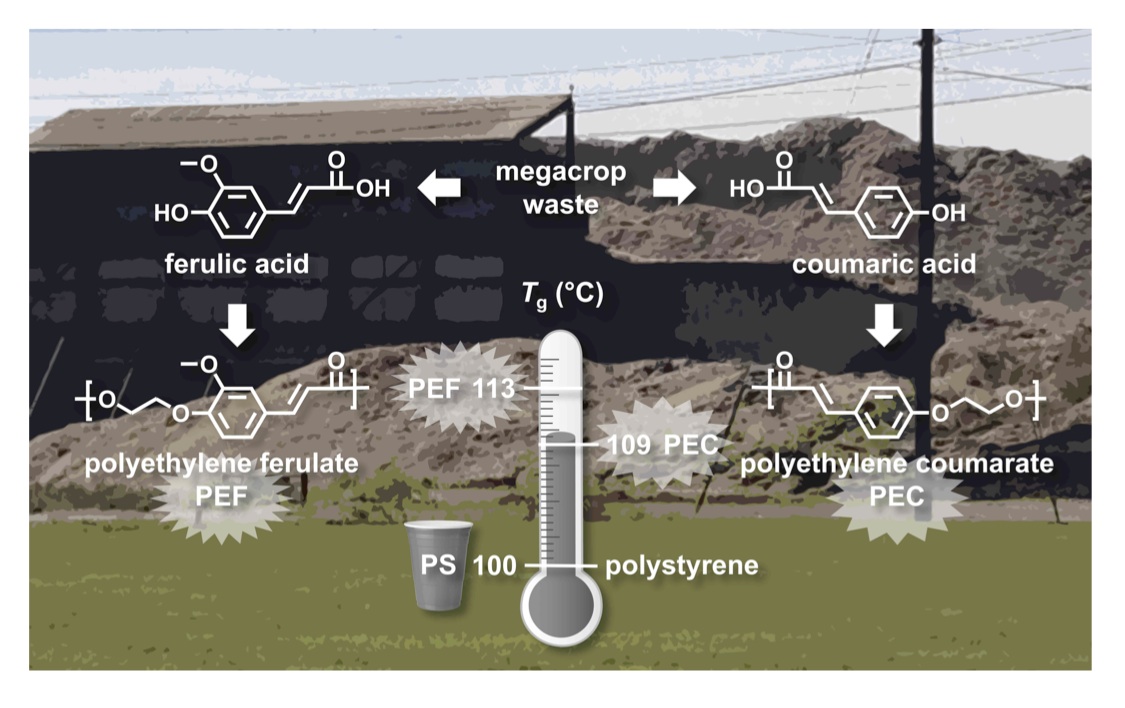
The hydrogenation of the double bond in ferulic acid is one approach for incorporating the necessary conformational flexibility into the polymer main-chain. An alternative strategy is to leave the double bond intact and install sp3 hybridized carbons elsewhere. Thus, the following four monomers were synthesized by appending ethylene spacers to ferulic acid and p-coumaric acid, an abundant, bioavailable cousin of ferulic acid: hydroxyethylferulic acid, hydroxyethyldihydroferulic acid, hydroxyethylcoumaric acid, and hydroxyethyldihydrocoumaric acid. The homopolymerization and copolymerization of these monomers yield a novel family of biorenewable thermoplastics with programmable glass transition temperatures. [14] For example, the polyethylene ferulate (PEF) homopolymer exhibited a Tg of 113 °C and the polyethylene dihydroferulate homopolymer exhibited a Tg of 32 °C. The corresponding copolymer series spanned the full range of intermediate Tg values, matching those of several high-volume commodity polymers. Importantly, the ferulic acid-based copolymer with a 10:90 feed ratio (predominantly composed of hydroxyethylferulic acid with 10% of the hydrogenated congener) exhibited a Tg of 98 °C, an excellent match for the Tg of polystyrene (PS). Similarly, the 10:90 coumaric acid-based copolymer showed a Tg of 94 °C, suggesting that it could also serve as a PS mimic. With promising properties and scalable feedstock availability, copolyesters from substituted hydroxycinnamic acids could prove to be sustainable replacements for a variety of non-renewable and non-degradable commodity plastics.
|
|
[1] Martin, R. T.; Camargo, L. P.; Miller, S. A. “Marine-Degradable Polylactic Acid”, Green Chem. 2014, 16, 1768–1773.
http://dx.doi.org/10.1039/C3GC42604A
Cover graphic for the April 2014 issue of Green Chemistry: http://dx.doi.org/10.1039/C4GC90011A
[2] Polyesteracetals. Inventors: Stephen A. Miller, Ryan T. Martin. U.S. Patent 8,653,226, issued February 18, 2014 (University of Florida).
http://patft.uspto.gov/netacgi/nph-Parser?patentnumber=8653226
[3] Pemba, A. G.; Flores, J. A; Miller, S. A. “Acetal Metathesis Polymerization (AMP): A method for synthesizing biorenewable polyacetals” Green Chem. 2013, 15, 325–329.
http://dx.doi.org/10.1039/C2GC36588J
[4] Acetal Metathesis Polymerization. Inventors: Stephen A. Miller, Alexander G. Pemba. U.S. Patent Application, PCT/US2012/029355; filed March 16th, 2012 (University of Florida, UF# 13521).
http://www.google.com/patents/WO2012129070A2?cl=en
[5] Miller, S. A. “Sustainable Polymers: Opportunities for the Next Decade” (Viewpoint), ACS Macro Lett. 2013, 2, 550–554.
http://dx.doi.org/10.1021/mz400207g
Journal Cover for ACS Macro Letters: http://pubs.acs.org/toc/amlccd/2/6
[6] Vanderhenst, R.; Miller, S. A. “Polycarbonates from Biorenewable Diols via Carbonate Metathesis Polymerization” Green Materials 2013, 1, 64–78.
http://dx.doi.org/10.1680/gmat.12.00022
[7] Garcia, J. J.; Miller, S. A. “Polyoxalates from Biorenewable Diols via Oxalate Metathesis Polymerization” Polym. Chem. 2014, 5, 955–961.
http://dx.doi.org/10.1039/C3PY01185B
[8] Sahmetlioglu, E.; Nguyen, H. T. H.; Nsengiyumva, O.; Göktürk, E.; Miller, S. A. “Silicon Acetal Metathesis Polymerization” ACS Macro Lett. 2016, 5, 466-470.
http://dx.doi.org/10.1021/acsmacrolett.6b00095
[9] Göktürk, E.; Pemba, A. G.; Miller, S. A. “Polyglycolic Acid from the Direct Polymerization of Renewable C1 Feedstocks” Polym. Chem. 2015, 6, 3918-3925.
http://dx.doi.org/10.1039/c5py00230c
Polymer Chemistry inside cover graphic: http://dx.doi.org/10.1039/C5PY90077H
[10] Poly(dihydroferulic acid): A biorenewable polyethylene terephthalate mimic derived from lignin and acetic acid and Copolymers Thereof. Inventors: Stephen A. Miller, Laurent Mialon. U.S. Patent 9,080,011, issued July 14, 2015 (University of Florida).
http://patft.uspto.gov/netacgi/nph-Parser?patentnumber=9080011
[11] PHFA technology licensed by U.S. Bioplastics. http://www.usbioplastics.com
[12] Mialon, L.; Pemba, A. G.; Miller, S. A. “Biorenewable polyethylene terephthalate mimics derived from lignin and acetic acid” Green Chem. 2010, 12, 1704–1706.
http://dx.doi.org/10.1039/C0GC00150C
Cover graphic for the October 2010 issue of Green Chemistry: http://dx.doi.org/10.1039/C0GC90024A
[13] Nguyen, H. T. H.; Suda, E. R.; Bradic, E. M.; Hvozdovich, J. A.; Miller, S. A. “Polyesters from Bio-Aromatics” ACS Symposium Series, Green Polymer Chemistry: Biobased Materials and Biocatalysis, Cheng, H. N.; Gross, R.; Smith, P., Eds., 2015, Vol. 1192, Chapter 24, 401–409.
http://dx.doi.org/10.1021/bk-2015-1192.ch024
Chapter graphic shown on rightmost hexagon of the cover: http://dx.doi.org/10.1021/bk-2015-1192
[14] Nguyen, H. T. H.; Reis, M. H.; Qi, P.; Miller, S. A. “Polyethylene ferulate (PEF) and congeners: polystyrene mimics derived from biorenewable aromatics”, Green Chem. 2015, 17, 4512-4517.
http://dx.doi.org/10.1039/c5gc01104c
Cover graphic for the September 2015 issue of Green Chemistry:
http://dx.doi.org/10.1039/c5gc90044A
|
Single-Site Catalysts for Olefin Polymerization
|
A small contingent of the Miller Research Group continues to pursue novel polyolefin structures, made possible by the unusual polymerization behavior of certain single-site olefin polymerization catalysts. In the recent past, this work has mostly been accomplished by post-docs, [15] visiting professors, [16] or visiting sponsored graduate student fellows, [17,18,19] with very targeted research objectives in the olefin polymerization area.
|
|
Catalysts with high alpha-olefin affinity
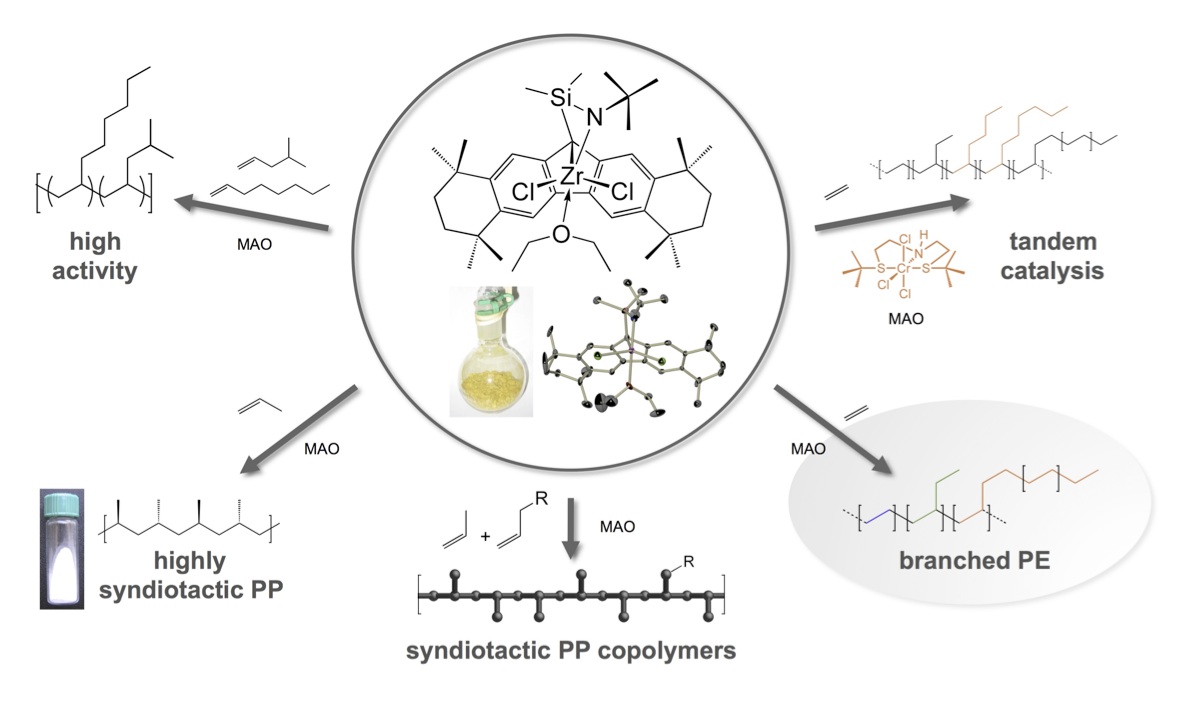
The commercial production of branched polyethylene is performed either by the classical high-temperature, high-pressure free radical process (low-density PE, LDPE), or at lower temperatures and pressures via the copolymerization of ethylene with alpha-olefins (linear low-density PE, LLDPE). In the latter case, the process conditions or comonomer add significant cost. Hence, a standing goal is to find a single-site catalytic system that can make branched polyethylene from ethylene alone. Our “Oct-Amido” zirconium-based catalyst exceeds all other early transition metal systems for the creation of branches (>50 branches/1000 carbons) by an order of magnitude; this is apparently accomplished via a macromonomer re-incorporation event that is very slow with “normal” catalysts. [20] However, molecular weights are likely too low for commercial utility and this downfall has been a prime target of our efforts, which include redesign of the organometallic catalyst, modification of the polymerization conditions, and inclusion of crosslinking agents. One way to exploit this molecular weight limitation is the oligomerization of alpha-olefins in the presence of hydrogen for preparing synthetic oils. Present work focuses largely on the copolymerization of ethylene and propylene, which exploits the unprecedented reactivity ratios of the Oct-Amido catalyst and other Oct-based catalysts. The high relative affinity of the “Oct-Cp” catalyst (which does make high Mn polyolefins) for alpha-olefins vs. ethylene (rethylene = 1.03; rpropylene = 0.64 at Tp = 0 °C) translates into the potential for new copolymer architectures, especially those from ethylene and propylene, the two most inexpensive hydrocarbon olefins.
|
|
[15] Chai, J.; Abboud, K. A.; Miller, S. A. “Sterically expanded CGC catalysts: Substituent effects on ethylene and alpha-olefin polymerization” Dalton Trans. 2013, 42, 9139–9147.
http://dx.doi.org/10.1039/C3DT50163A
[16] Zohuri, G. H.; Albahily, K.; Schwerdtfeger, E. D.; Miller, S. A. Metallocene Alkene Polymerization Catalysts, In Polymer Science: A Comprehensive Reference, Vol. 3; Matyjaszewski, K.; Möller, M., Eds.-in-Chief; Elsevier: Amsterdam, 2012; pp. 673–697.
http://dx.doi.org/10.1016/B978-0-444-53349-4.00081-9
[17] Nejabat, G. R.; Nekoomanesh, M.; Arabi, H.; Salehi-Mobarakeh, H.; Zohuri, G. H.; Omidvar, M.; Miller, S. A. “Synthesis of Stereoblock Elastomeric Poly(propylene)s Using a (2-PhInd)2ZrCl2 Metallocene Catalyst in the Presence of Co-Catalyst Mixtures, 1–Study of Activity and Molecular Weight” Macromol. React. Eng. 2012, 6, 523–529.
http://dx.doi.org/10.1002/mren.201200046
[18] Nejabat, G. R.; Nekoomanesh, M.; Arabi, H.; Salehi-Mobarakeh, H.; Zohuri, G. H.; Omidvar, M.; Miller, S. A. “Synthesis and Microstructural Study of Stereoblock Elastomeric Polypropylenes from Metallocene Catalyst (2-PhInd)2ZrCl2 Activated with Cocatalyst Mixtures” J. Polym. Sci. Part A: Polym. Chem. 2013, 51, 724–731.
http://dx.doi.org/10.1002/pola.26432
[19] Nejabat, G.-R.; Nekoomanesh, M.; Arabi, H.; Salehi-Mobarakeh, H.; Zohuri, G.-H.; Mortazavi, S. M. M.g; Ahmadjo, S.; Miller, S. A. “Study of Ziegler-Natta/(2-PhInd)2ZrCl2 hybrid catalysts performance in slurry propylene polymerization”, Polyolefins Journal 2015, 2, 73–87.
http://poj.ippi.ac.ir/article_1145_341.html
[20] Schwerdtfeger, E. D.; Irwin, L. J.; Miller, S. A. "Highly Branched Polyethylene from Ethylene Alone via a Single Zirconium-Based Catalyst" Macromolecules 2008, 41, 1080–1085.
http://dx.doi.org/10.1021/ma702213c
|
Polymer/Metal-Organic-Frameworks (polyMOFs)
|
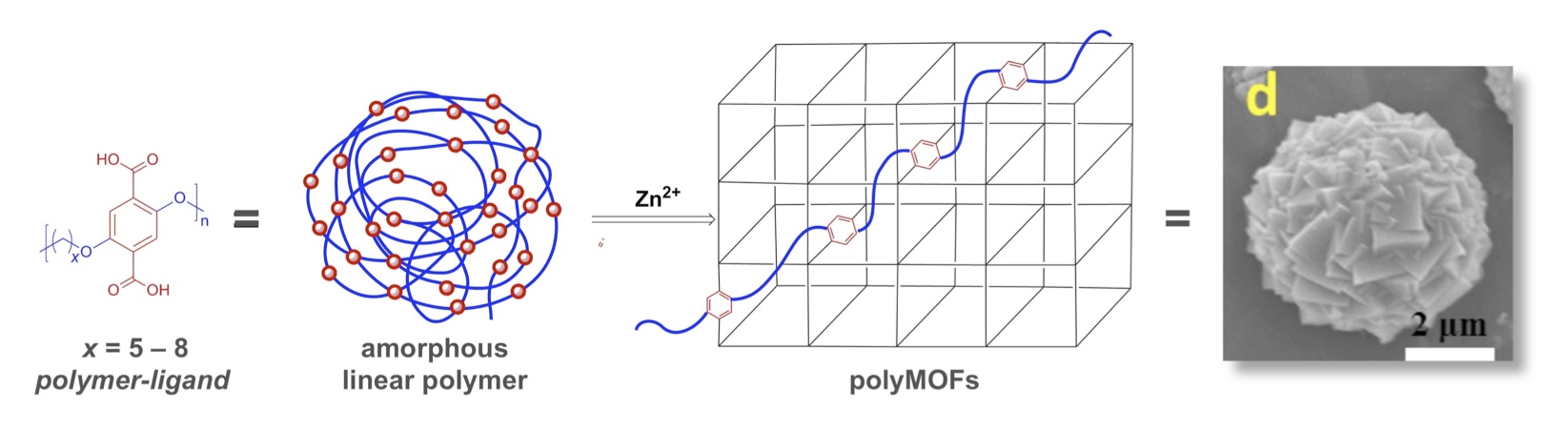
A recent collaboration with Prof. Seth Cohen at the University of California, San Diego (UCSD) has resulted in a new class of porous materials, located at the intersection of polymer chemistry and metal-organic-frameworks. Preparation of porous one-dimensional polymers is challenging because the dense and efficient packing of polymer chains results in non-porous, amorphous materials. The Miller Research Group (polymer chemistry) and the Cohen Research Group (coordination chemistry) have demonstrated the remarkable transformation of a series of linear, non-porous, largely amorphous, organic polymers into three-dimensional, highly-porous, crystalline, metal-organic framework (MOF) hybrid materials. [21,22,23] This was achieved with an unprecedented design and synthesis strategy: first, preparing polymer-ligands containing multiple terephthalate moieties; and second, annealing the polymer-ligands with Zn(II) ions to generate a new hybrid material we have termed a polymer-metal-organic-framework (polyMOF). Key findings include: 1) The first demonstration that a linear, organic polymer can be readily reorganized into a highly crystalline, porous, three-dimensional framework structure directed by coordination chemistry; 2) The first example of preparing crystalline polymer-MOF hybrid materials using a bottom-up synthesis. These novel materials uproot the dogma that MOFs can only be prepared with relatively small, rigid, organic ligands; 3) The potential to harness the advantages of both polymers and MOFs, including moisture resistance and permanent porosity. Moreover, some polyMOFs exhibit enhanced CO2 uptake and isosteric heat of CO2 adsorption; 4) polyMOF materials exhibit novel and tunable morphologies, including crystalline, spherical superstructures, and films. The Miller Research Group and the Cohen Research group are vigorously pursuing this collaboration, which included a two-week sabbatical taken by Professor Cohen at the University of Florida in September, 2015.
|
|
[21] Zhang, Z.; Nguyen, H. T. H.; Miller, S. A.; Cohen, S. M. “polyMOFs: A Class of Interconvertible Polymer-Metal-Organic-Framework Hybrid Materials” Angew. Chem. Int. Ed. 2015, 54, 6152–6157.
http://dx.doi.org/10.1002/anie.201502733
Angewandte Chemie Frontispiece Graphic: http://dx.doi.org/10.1002/anie.201582161
[22] Chemical & Engineering News Research Editorial “MOFs Made From Flexible Polymers Buck Conventional Wisdom”, by Mitch Jacoby, May 7, 2015.
http://cen.acs.org/articles/93/i19/MOFs-Made-Flexible-Polymers-Buck.html
[23] Zhang, Z.; Nguyen, H. T. H.; Miller, S. A.; Ploskonka, A. M.; DeCoste, J. B.; Cohen, S. M. “polyMOFs as Water Tolerant Materials for Selective Carbon Dioxide Separations” J. Am. Chem. Soc., Accepted December 28, 2015.
http://dx.doi.org/10.1021/jacs.5b11034
|
|
|
| |
|